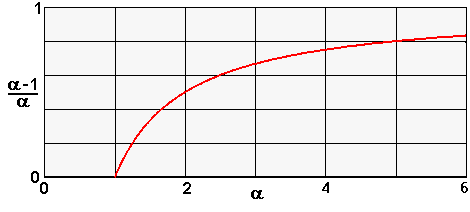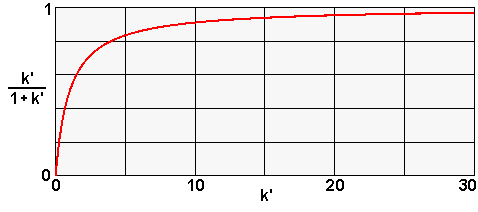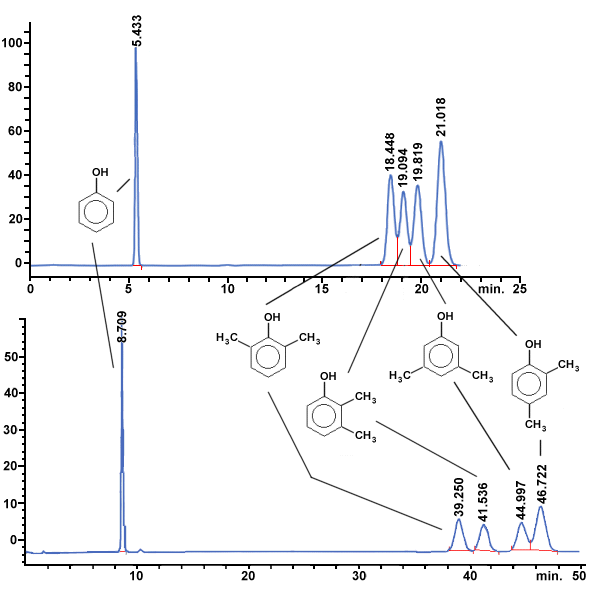

| La valeur de K dépend - de la structure du composé qui détermine son affinité pour chacune des phases | |
| - de la nature de la phase stationnaire qui est un adsorbant ou un solvant pour chacun des composés | |
| - de la phase mobile, seulement si elle est un liquide, et donc un solvant pour chacun des composés | |
| - de la température qui affecte les pressions de vapeur et les solubilités | |
| Chromatographie en phase gazeuse | ||
| type | phase stationnaire | |
| gaz/solide | adsorption | solide poreux |
| gaz/liquide | partage (partition) |
dans les
colonnes remplies, solide poreux inerte enrobé de liquide |
| dans les
colonnes capillaires, paroi interne de la colonne qui sert de support | ||
| Chromatographie en phase liquide | ||
| type | phase stationnaire | |
| liquide/solide | adsorption | solide poreux |
| échange d'ions | solide à la surface duquel se trouvent des sites ioniques qui permettent à l'aide d'un solvant approprié l'échange d'ions présents dans la phase mobile | |
| d'exclusion (filtration sur gel, perméation de gel) |
solide dont la dimension des pores permet la séparation des espèces selon leur taille | |
| liquide/liquide | partage phase normale |
solide poreux inerte enrobé de liquide (de moins en moins utilisée) |
| partage phase inversée |
solide poreux sur lequel sont greffées des chaînes hydrocarbonées non-polaires | |
| Chromatographie en phase supercritique | |
| gaz supercritique/solide | |
| gaz supercritique/liquide | |


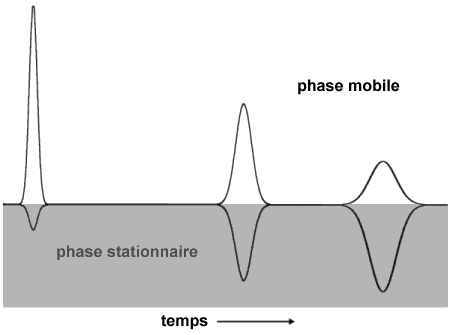

b) le composé est absorbé trop rapidement dans la phase stationnaire, il migre trop vite et laisse un effet de queue; c) le composé est absorbé trop lentement dans la phase stationnaire, sa migration est trop lente. |


| Le temps de rétention est habituellement utilisé à la place du volume de rétention et sa grandeur dépend: | |
| - de la nature de la phase stationnaire | |
| - de la nature de la phase mobile | |
| - du débit de la phase mobile | |
| - de la longueur de la colonne | |

| où tm | = temps de rétention d’une substance non retenue sur la colonne | |
| tr | = temps de rétention | |
| t’r | = temps de rétention corrigé par rapport à tm | |
| l | = largeur du pic à la base | |
| h | = hauteur du pic | |
| h’ | = hauteur corrigée du pic |
| Figure 1.5. Chromatogramme typique montrant la séparation de deux constituants d’un mélange et les différents paramètres mesurables. |
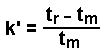


| Figure 1.6. a) les pics se
recouvrent; b) la distance séparant les sommets des pics est plus grande mais les largeurs à la base sont les mêmes qu’en a); c) la distance séparant les sommets des pics est égale à celle trouvée en a) mais les largeurs à la base sont plus petites qu’en b). |
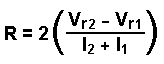
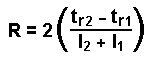
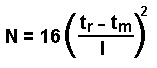 |
où l = largeur du pic à la base |
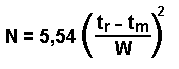 |
où w = largeur du pic à mi-hauteur |
| La hauteur équivalente à un plateau théorique h est | |
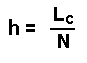 |
où LC =
longueur de la colonne et N = nombre de plateaux théoriques |