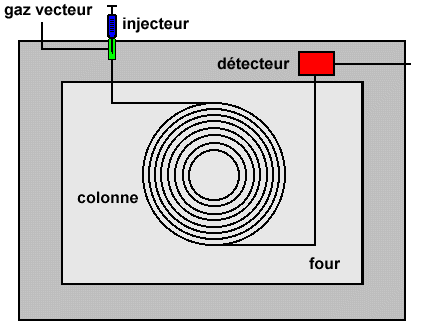
| Cette opération est faite - à l’aide d’une micro seringue - pour les liquides et les solutions | |
| - à l’aide d’une vanne à boucles - pour les mélanges gazeux | |

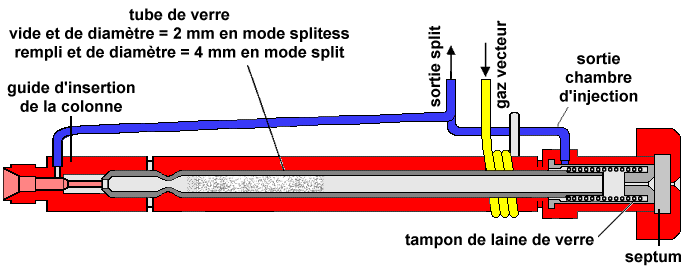
 |
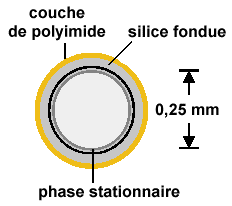 | |
| Figure 2.4. Colonne
remplie |
Figure 2.5. Colonne capillaire. | |
| Les représentations ne sont pas à l’échelle entre colonnes remplie et capillaire. |
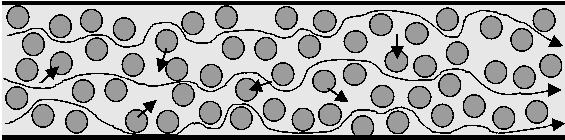
| Figure 2.6. |
| Figure 2.7. |
| Quelques caractéristiques des colonnes remplies et colonnes capillaires | |||
| diamètre
interne (mm) |
capacitéa (ng) |
plateaux théoriques par mètre |
débit
optimal (mL/minute) |
| 0,20 | 5-30 | 5000 | 0,40 |
| 0,25 | 50-100 | 4170 | 0,70 |
| 0,32 | 400-500 | 3330 | 1,40 |
| 0,53 | 1000-2000 | 1670 | 2,50 |
| 0,75 | 10000-15000 | 1170 | 5,00 |
| 2,00b | 20000- | 2000 | 20,00 |
| a capacité en fonction de l’épaisseur
de la couche de la phase stationnaire b colonne remplie | |||
| Molécules | Exemples | Phases stationnaires |
| non-polaires liaisons C-H et C-C |
hydrocarbures
normaux (n-alcanes) |
SPB-octyl |
| poly(méthylsiloxane), SE-30 | ||
| SPB-5, PTE-5, SE-54 | ||
| polaires liaisons C-H et C-C liaisons C-Cl, -Br, -F liaisons C-N, -O, -P, -S |
alcools, éthers,
thiols, amines, acides carboxyliques, esters et cétones |
poly(méthylphénylsiloxane) |
| poly(cyanopropylméthylsiloxane) | ||
| PEG, Carbowax 10, 20M | ||
| polarisables liaisons C-H et C=C et acétyléniques |
alcènes aromatiques | poly(cyanopropylsiloxane) |
| poly(cyanopropylphénylsiloxane) | ||
| TCEP |

| Figure 2.13. Détecteur à conductibilité thermique. |
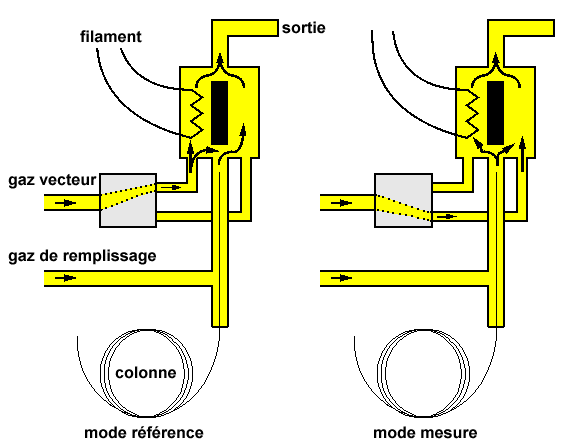
| Figure 2.14. Sur le schéma de gauche, le gaz vecteur est dirigé sur la filament. À la sortie de colonne, le gaz vecteur contenant le soluté est envoyé directement vers la sortie. Sur celui le schéma de droite, le gaz vecteur est envoyé vers la sortie et le gaz vecteur contenant le soluté est dirigé vers la filament. |
| Conductibilité thermique par rapport à l’azote |
à 100oC |
| Hydrogène | 6,95 |
| Hélium | 5,54 |
| Méthane | 1,72 |
| Azote | 1,00 |
| Argon | 0,71 |
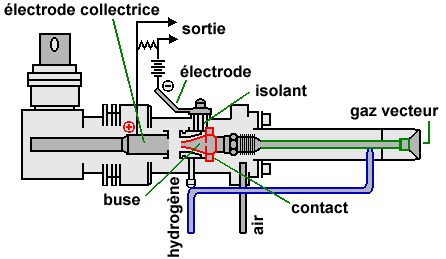
| Figure 2.15. Détecteur à ionisation de flamme du modèle 8310 de la compagnie Perkin-Elmer. |

| Figure 2.16. Couplage entre le chromatographe en phase gazeuse et le spectromètre de masse par un séparateur à jet moléculaire chauffé pour éviter la condensation des substances éluées. |

