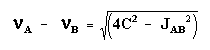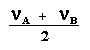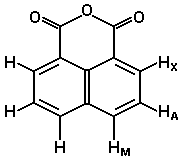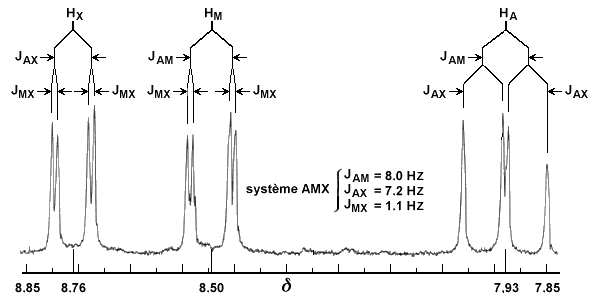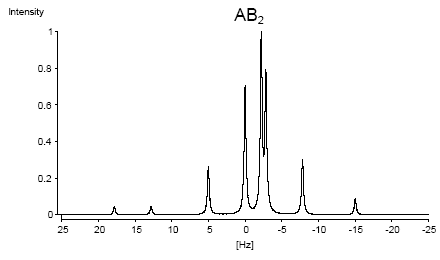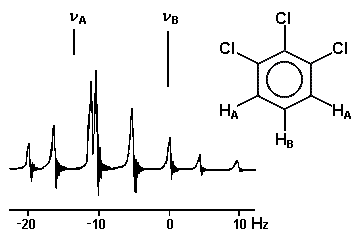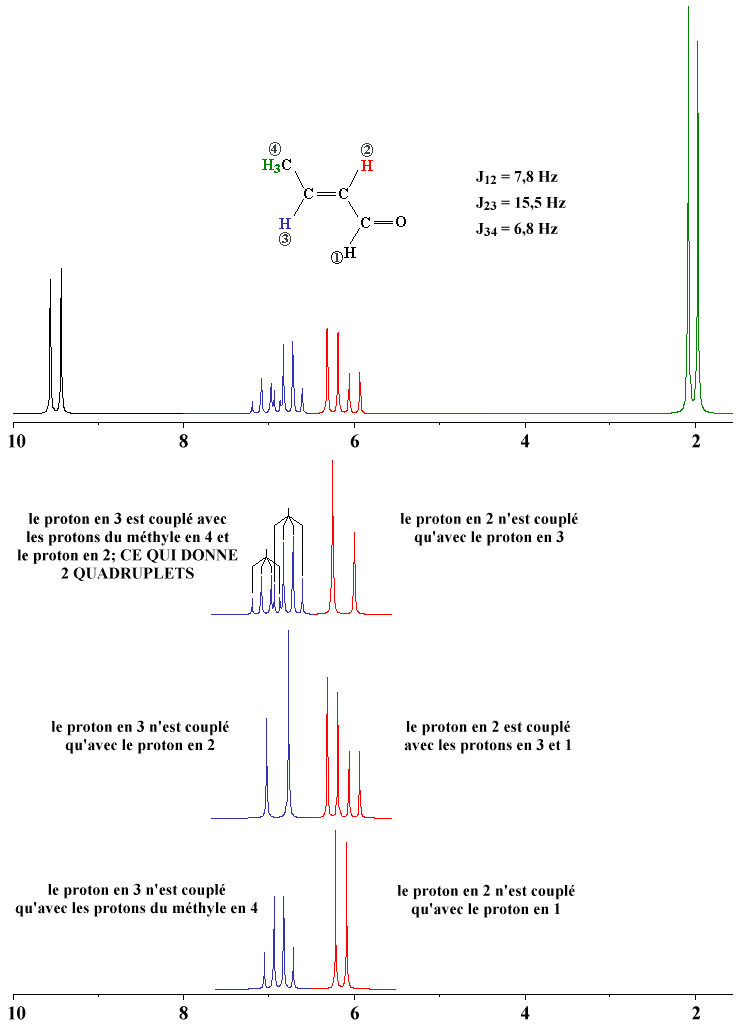

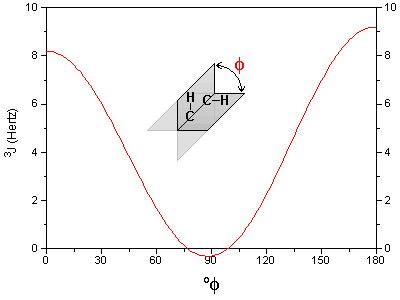
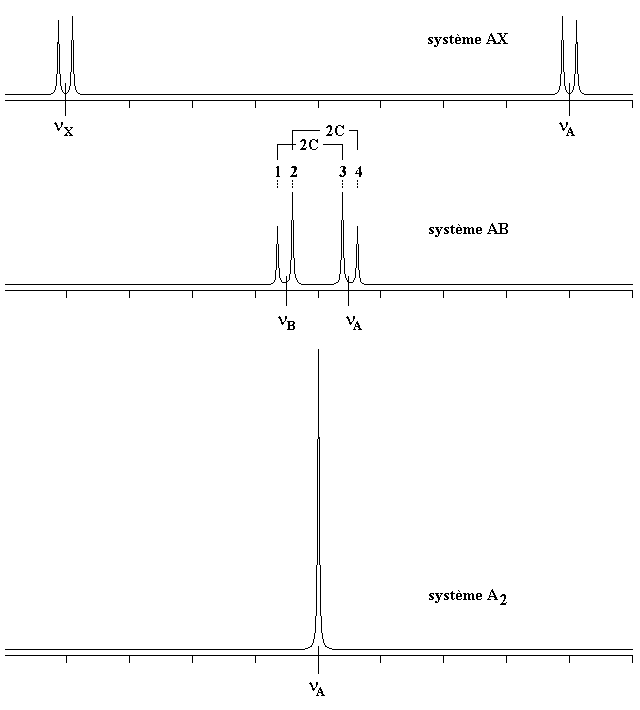
Le cas AB est le plus simple de ces systèmes. L'aspect du spectre pour le cas AB est intermédiaire entre ceux des cas AX et A2. Le cas AX peut être résolu à partir des mesures faites directement sur le spectre. Le système A2 est encore plus simple puisqu'il ne comprend qu'une seule bande. |